Generalidades de la Diabetes Insípida Central y Nefrogénica
Overview of Central and Nephrogenic Diabetes Insipidus
Dra. María Paula Pacheco Salas
Médico y Cirujano
Investigadora independiente, San José, Costa Rica.
Esta dirección de correo electrónico está siendo protegida contra los robots de spam. Necesita tener JavaScript habilitado para poder verlo.
https://orcid.org/0000-0003-2282-4513
Dra. Amanda Coto Quirós
Médico y Cirujano
Investigadora independiente, San José, Costa Rica.
Esta dirección de correo electrónico está siendo protegida contra los robots de spam. Necesita tener JavaScript habilitado para poder verlo.
https://orcid.org/0000-0002-3395-9184
Dr. Manrique Sauma Montes de Oca
Médico y Cirujano
Investigador independiente, San José, Costa Rica.
Esta dirección de correo electrónico está siendo protegida contra los robots de spam. Necesita tener JavaScript habilitado para poder verlo.
https://orcid.org/0000-0002-5172-280X
Dr. Kevin Villarevia Umaña
Médico y Cirujano
Investigador independiente, San José, Costa Rica
Esta dirección de correo electrónico está siendo protegida contra los robots de spam. Necesita tener JavaScript habilitado para poder verlo.
https://orcid.org/0000-0001-8810-1807
Pacheco-Salas, M.; Coto-Quirós, A.; Sauma-Montes de Oca, M.; Villarrevia-Umana, K. Revisión Bibliográfica: Generalidades de la Diabetes Insípida Central y Nefrogénica. Crónicas Científicas. Vol.24. No.24. Pág. 27-36 ISSN: 2215-5171
Fecha de recepción: Febrero 2023
Fecha de aceptación: Agosto 2023
Resumen
La diabetes insípida (DI) es una enfermedad caracterizada por poliuria y polidipsia. Puede ser de origen central o nefrogénica, y en casos raros inducida por el embarazo. La DI central resulta de la deficiencia hormona antidiurética (ADH), mientras la DI nefrogénica resulta de la resistencia renal a la ADH. En ambos casos, las causas adquiridas son mucho más comunes que las causas genéticas o hereditarias. Es una enfermedad poco frecuente, con prevalencia de 1:25 000, que no tiene predilección por sexo y se puede presentar a cualquier edad. Debido a que los síntomas son los mismos que para polidipsia primaria, diferenciar entre ambas conlleva un reto diagnóstico. Clásicamente se utiliza el test de deprivación de agua de dos pasos. La capacidad de medición de copeptina por sí misma y posterior a infusión de solución salina hipertónica han mejorado la precisión diagnóstica de la enfermedad. Se debe buscar la causa de la enfermedad con base en la sospecha diagnóstica, ya que dar un tratamiento a un diagnóstico errado puede tener consecuencias graves. La base del tratamiento farmacológico de DI nefrogénica son los diuréticos tiazídicos, mientras que para la DI central es la desmopresina, un análogo de la ADH.
Palabras claves
Diabetes insípida, diabetes insípida nefrogénica, diabetes insípida central, síndrome poliuria polidipsia.
Abstract
Diabetes insipidus (DI) is a disease characterized by polyuria and polydipsia. It can occur due to antidiuretic hormone (ADH) deficiency in central DI; due to a renal resistance to ADH in nephrogenic DI, and in rare cases induced by pregnancy. Causes of both are usually acquired. It is a rare disease, with a prevalence of 1:25000, which has no predilection by sex and can be present at any age. Symptoms for DI and primary polydipsia are the same, so distinguishing between both can be challenging. Classically, the two-step water deprivation test is used. The ability to measure copeptin by itself and its measure after an infusion of hypertonic solution have improved diagnostic accuracy of the disease. An appropriate diagnostic and its cause should be done based on clinical suspicion, since misleading the diagnosis and treating it wrongly can have serious consequences. The mainstay of drug treatment for nephrogenic DI is thiazide diuretics, while for central DI it is desmopressin, an ADH analogue.
Keywords
Diabetes insipidus, nephrogenic diabetes insipidus, central diabetes insipidus, poliuria-polydipsia syndrome.
Método
Esta revisión bibliográfica se realizó mediante una búsqueda en bases de datos de UpToDate, Pubmed y el Sistema de Bibliotecas, Documentación e Información de la Universidad de Costa Rica. Para ello, se utilizaron criterios de inclusión mediante búsquedas relacionadas a “Síndrome Poliuria Polidipsia”, “Diabetes Insípida”, “Diabetes Insípida central”, “Diabetes Insípida Nefrogénica”. Se utilizaron 17 artículos en el idioma original inglés y con antigüedad máxima de 5 años, comprendiendo un periodo de 2019 - 2023.
Introducción
La diabetes insípida (DI) es una enfermedad que forma parte del síndrome de poliuria-polidipsia. Se caracteriza por poliuria hipotónica (excreción urinaria de más de 50mL/ kg/24h) y polidipsia (ingesta de más de 3L/día).1
Las formas más frecuentes de DI son la central y la nefrogénica. La forma central ocurre por una secreción insuficiente de hormona antidiurética (ADH) o vasopresina en el hipotálamo. La DI nefrogénica ocurre por una respuesta renal reducida a niveles fisiológicos de hormona antidiurética. En la mayoría de casos, tanto la forma central como la nefrogénica son adquiridas, sin embargo, también existen formas congénitas que se revisarán más adelante. Es importante al momento de sospechar el diagnóstico, tomar en cuenta formas secundarias de poliuria que pertenecen al síndrome poliuria polidipsia, por ejemplo, polidipsia primaria.2
Existe una tercera forma de la enfermedad, la DI gestacional, que es aún menos frecuente y ocurre por aumento del metabolismo enzimático (por la vasopresinasa placentaria) de la ADH durante el embarazo.1,3
Objetivo
El objetivo de esta revisión bibliográfica es sintetizar de fuentes bibliográficas recientes la epidemiología, fisiopatología, manifestaciones clínicas, diagnóstico y tratamiento de la diabetes insípida, tanto central como nefrogénica. Así como brindar conclusiones que puedan ser útiles para la práctica de medicina general en cuanto al conocimiento de esta enfermedad.
Epidemiología
La DI es una enfermedad poco frecuente, con una prevalencia de 1:25 000, que no tiene predilección en cuanto al sexo. Se puede presentar a cualquier edad dependiendo de su etiología.2 Las formas hereditarias se van a presentar más tempranamente, mientras que las formas adquiridas suelen presentarse posterior a la temprana infancia. Las formas adquiridas de la enfermedad son mucho más comunes, menos de 10% de los casos de DI central o nefrogénica se deben a causas hereditarias.1
La forma más común de DI es la central, en la mayoría de los casos debido a causas adquiridas por daño en las células productoras de vasopresina en la neurohipófisis.3
En cuanto a la DI nefrogénica adquirida, se debe hasta en un 30% al uso crónico de tratamiento con litio. Sobre las causas hereditarias de DI nefrogénica, se ha estimado en algunos estudios un 90% de casos debido a mutaciones en el gen AVPR2.1
Fisiopatología
La ADH o vasopresina se produce en el núcleo supraóptico y paraventricular del hipotálamo, posteriormente es secretada por la glándula pituitaria o hipófisis en respuesta al aumento en la osmolalidad plasmática o hipovolemia. El umbral para la secreción de ADH es 280 – 290mOsm/ kg, al haber un aumento por encima de este umbral se estimula la secreción de la misma.4
DI NEFROGÉNICA
La ADH tiene su acción a nivel de los receptores V2 en la membrana basolateral de las células principales del túbulo colector y la región ascendente gruesa del asa de Henle en el riñón. Una vez que se da la unión estos receptores se estimula la producción intracelular de adenosin monofosfato cíclico (AMPc), que a su vez activa la protein kinasa dependiente de AMPc. La proteína se encarga de la fosforilación y translocación de las acuaporinas 2 (AQP2) a la membrana apical del túbulo colector. De esta manera, se permite la difusión de agua a través de las células principales y el intersticio, lo cual genera una mayor reabsorción de agua a través de la médula renal por gradiente de concentración. Favoreciendo como resultado final la capacidad de concentración de orina.4, 5
La DI nefrogénica es causada por insensibilidad renal a niveles fisiológicos de ADH, generando poliuria y consecuentemente lleva a polidipsia. De las formas congénitas, lo más frecuente es que ocurran mutaciones en el gen AVPR2, pero también pueden ocurrir mutaciones del canal de AQP2.4 El gen AVPR2 que codifica para una proteína G que se acopla al receptor hormonal V2, se localiza en el cromosoma X, por lo que las mutaciones en el mismo van a ser ligadas al X y generalmente afecta individuos de sexo masculino.6, 7
La DI nefrogénica adquirida es debida principalmente a medicamentos, dentro de los cuales destacan aminoglicósidos, foscarnet, rifampicina, furosemida, colchicina y más comúnmente el litio. La acumulación citotóxica de Litio en las células lleva a una disminución de la expresión de AQP2.8 Su fisiopatología también se ha visto asociada con anormalidades electrolíticas y uropatías obstructivas.4
DI CENTRAL
El mecanismo fisiopatológico común de la DI central es una producción o secreción insuficiente de ADH. La mayoría de los casos de DI central son adquiridos por daño en las células productoras de ADH, por ejemplo: trauma, cirugía, causas vasculares o enfermedades granulomatosas.3
Existen además un grupo enfermedades congénitas que se han asociado con DI central, dentro de ellas la DI central familiar y el Síndrome de Wolfram. La DI central familiar es una enfermedad autosómica dominante causada por mutaciones en el gen que codifica para la ADH. A pesar de ser una enfermedad autosómica dominante, la enfermedad en individuos afectados puede pasar desapercibida, debido a que se puede producir ADH funcional a partir del alelo que no está afectado, y el retículo endoplásmico degrada la ADH que tiene la mutación, por lo que la enfermedad puede no tener manifestaciones clínicas.9
El síndrome de Wolfram es un síndrome caracterizado por DI central, diabetes mellitus, atrofia óptica y sordera, con manifestaciones cognitivas o psiquiátricas que aparecen con el desarrollo de la enfermedad. Es causado por defecto de al menos dos genes, el WFS1 y CISD2, que codifican para proteínas del retículo endoplásmico que afectan la homeostasis del Calcio. La DI en este síndrome se presenta debido a una pérdida de neuronas secretoras de ADH en el núcleo supraóptico y procesamiento inadecuado de los precursores de ADH.9
Presentación Clínica
Los síntomas pivote en DI son poliuria (excreción urinaria de más de 40-50mL/kg/día) y polidipsia (ingesta de agua de más de 100mL/kg/día). Estos síntomas pueden presentarse de manera gradual o abrupta, dependiendo de la etiología de la enfermedad. Los pacientes además pueden reportar nicturia, fatiga, dificultad para dormir y pérdida de peso.5 El inicio abrupto de los síntomas, la persistencia de los síntomas durante la noche y la preferencia por bebidas frías son típicos de DI y son elementos de la historia que pueden ayudar a diferenciarlo de otras etiologías del síndrome de poliuria-polidipsia como la polidipsia primaria. Sin embargo, existen etiologías de DI central que suelen presentar síntomas de manera gradual o larvada, como síndromes inflamatorios, post radiación o por mutaciones en el gen que codifica para ADH.10
Debido a que los síntomas pueden estar presentes tanto en DI como polidipsia primaria, suele ser difícil diferenciarlas clínicamente. Además, si bien las comorbilidades psiquiátricas suelen ser más comunes en polidipsia primaria, también se han observado en DI con menor frecuencia.10
En niños, se pueden presentar síntomas como frecuencia urinaria, nicturia, enuresis, sed e irritabilidad. En casos de DI severa existe riesgo de presentar deshidratación severa y falla para progresar.5
La historia clínica y el examen físico se deben dirigir de manera que se puedan identificar elementos sugestivos de enfermedad intracraneal, trauma o cirugía reciente, déficits hormonales hipofisiarios concomitantes, enfermedades renales u otras enfermedades sistémicas. Así como identificar ingesta de medicamentos que puedan estar relacionados con DI.5
Diagnóstico
Para el diagnóstico de DI se pueden seguir los siguientes pasos: 1) confirmar la poliuria 2) confirmar que la poliuria sea hipotónica 3) determinar el tipo de DI 4) determinar la causa de DI.5
1. CONFIRMAR POLIURIA
Poliuria se define como excreción de más de 40-50mL/kg/ día de orina. Una vez confirmada la misma se puede continuar con los otros pasos diagnósticos.5
2. CONFIRMAR POLIURIA HIPOTÓNICA
Se debe medir la osmolalidad de la orina, la DI se debe sospechar en caso de que la orina tenga una osmolalidad menor a 300mOsm/kg. Si la osmolalidad urinaria es mayor a 800mOsm/kg se deben considerar otras causas de diuresis osmótica como estados hiperglicémicos, diabetes mellitus, enfermedad renal, estados catabólicos, alta ingesta de proteínas, uso de medicamentos como diuréticos o manitol.5
La presencia poliuria hipoosmolar en presencia de hiperosmolalidad plasmática (más de 300mOsm/kg) e hipernatremia (más de 145mEq/L) son sugestivos de DI. Por el contrario, en polidipsia primaria ambos parámetros son normales o bajos.5
Clásicamente se utiliza el test de deprivación de agua para diferenciar DI de polidipsia primaria. En individuos con polidipsia primaria o sanos, la respuesta fisiológica a la deprivación de agua es un estímulo en la secreción de ADH, que lleva consecuentemente a un aumento en la osmolalidad urinaria de más de 800mOsm/kg. Por el contrario, en pacientes con DI no hay capacidad de concentrar la orina, de manera que la misma permanece diluida (con osmolalidad menor a 300mOsm/kg).5 Estos valores de corte para el test de deprivación de agua fueron determinados con una cohorte pequeña de pacientes, y en nuevos estudios se ha demostrado una baja precisión diagnóstica, siendo todavía más baja para diagnóstico de polidipsia primaria, por lo que se han propuesto otros elementos como la medición de copeptina para aumentar la precisión diagnóstica.11 Es necesario hacer énfasis en la importancia de diferenciar DI de polidipsia primaria, debido a que un diagnóstico erróneo y tratamiento con desmopresina a individuos con polidipsia primaria, puede tener efectos adversos graves como intoxicación por agua.3
3. DETERMINAR EL TIPO DE DI
En el algoritmo clásico, posterior a la finalización del test de deprivación de agua, los pacientes que persisten con osmolalidad urinaria menor a 300mOsm/kg continúan con la segunda parte del test. Este segundo paso consiste en administrar desmopresina intramuscular o subcutánea, con el objetivo de diferenciar DI central y nefrogénica. En DI central con la administración de la hormona, se espera que haya respuesta renal y la osmolalidad urinaria aumente, mientras que en DI nefrogénica la osmolalidad urinaria se mantiene baja.12
La medición de los niveles de ADH al finalizar el test de deprivación de agua (antes de administrar vasopresina) también se ha utilizado clásicamente en el algoritmo diagnóstico de DI. Los resultados esperados son: que sean bajos en DI central, altos en DI Nefrogénica y normales o bajos en polidipsia primaria o DI central parcial. Sin embargo, la ADH es un péptido pequeño que se metaboliza rápidamente por lo que su medición no es siempre fidedigna.5 La copeptina es un péptido que se secreta junto a la ADH, cuya función fisiológica no se tiene clara, que es mucho más estable que la hormona, por lo que su medición ha probado tener alta precisión diagnóstica en DI.5, 11
Los niveles de copeptina basales se han utilizado para el diagnóstico de DI y diferenciar si es central o Nefrogénica. Niveles basales < 2.6pmol/L es diagnóstico de DI central, mientras que niveles basales de > 21.4pmol/L es diagnóstico de DI nefrogénica. Si los niveles son indeterminados, se sugiere DI central o polidipsia primaria, en este caso es necesario medir la copeptina posterior a una infusión de solución hipertónica (250mL en 15min, seguido de una infusión a 0.15mL/g/min hasta alcanzar natremia de 150mEq/L). En individuos sanos o polidipsia primaria este estímulo va a producir la liberación de ADH, por lo que niveles de > 4.9 pmol/L indican polidipsia primaria y niveles < 4.9 pmol/L son sugestivos de DI central. La medición de niveles de copeptina han demostrado aumentar la precisión diagnóstica de la enfermedad.5, 11
4. DETERMINAR LA CAUSA DE DI
Para determinar la causa de DI, se tiene que tomar en cuenta el amplio diagnóstico diferencial de tanto DI central y DI nefrogénica, tomando en cuenta que en ambos casos las formas adquiridas son más frecuentes.4
DI NEFRO GÉNICA
Para el diagnóstico de DI nefrogénica en niños, se tiene que tener en cuenta dentro del diagnóstico diferencial las causas genéticas o hereditarias, que en un 90% se deben a una pérdida de función del receptor V2 debido a una mutación.4
En adultos, las formas adquiridas de DI nefrogénica son mucho más comunes. Se deben buscar elementos en la historia clínica, examen físico y laboratorios que hagan sospechar causas secundarias. Entre ellas se debe considerar: disfunción tubular, trastornos hidroelectrolíticos como hipercalcemia, hipokalemia, uropatía obstructiva, además de descartar uso de medicamentos previamente mencionados que pueden causar DI nefrogénica.4
DI CENTRAL
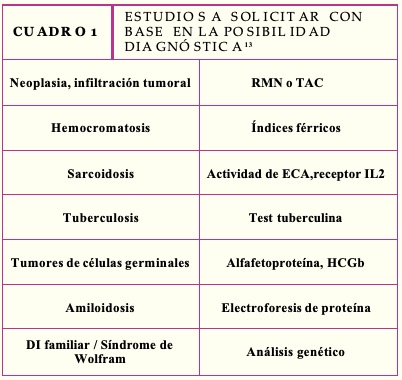
Se debe tomar en cuenta la rapidez de instauración de los síntomas, si es aguda o gradual, realizar una historia exhaustiva sobre otros síntomas como cefalea, anormalidades visuales, síntomas B, exposición a medicamentos o tóxicos, historia familiar de polidipsia, historia personal de trauma o cirugía reciente.13
Para evaluar las causas de DI central, se deben hacer laboratorios generales iniciales como hemograma, química incluyendo sodio, potasio, calcio, osmolalidad plasmática y glucosa. Con base en la sospecha diagnóstica se pueden solicitar laboratorios dirigidos y valorar la necesidad de estudios de imagen.13
Tratamiento
DI NEFRO GÉNICA
En niños el tratamiento de DI nefrogénica puede ser difícil, por lo que se les debe ofrecer líquidos cada 2h, con el objetivo de mantener una adecuada hidratación. En algunos casos se debe considerar la alimentación por sonda nasogástrica o gastrostomía, sobre todo en las noches. Otro aspecto importante sobre el tratamiento no farmacológico es una dieta con restricción en sodio y proteínas. La excreción de orina se determina por la carga osmótica de la misma, de manera que, al disminuir la carga osmótica con una restricción de solutos, se logra una disminución en la excreción urinaria.4
El tratamiento inicial de DI nefrogénica suele ser con diuréticos tipo tiazidas. Estos medicamentos bloquean el cotransportador de sodio-cloruro (NCC) en el túbulo contorneado distal, aumentando la concentración de sodio y osmolalidad urinaria. Este aumento en la natriuresis, disminuye el volumen intravascular potenciando el estímulo del sistema renina-angiotensina-aldosterona (SRAA), por lo que se disminuye el volumen de filtrado glomerular. Finalmente, la reabsorción de sodio y agua aumentan en el túbulo proximal, disminuyendo el volumen que llega a la parte distal de la nefrona y disminuyendo el volumen de orina. En niños se inicia típicamente con hidroclorotiazida a dosis de 2-4mg/kg/día dividido en 2 dosis, en adultos se utiliza 25mg una o dos veces al día.4, 14
La hipokalemia es un efecto secundario conocido de los diuréticos tiazidas, de manera que su combinación con un diurético ahorrador de potasio como amiloride, ha probado ser beneficioso. Sus beneficios también se han demostrado en DI nefrogénica inducida por litio, debido a que bloquea los ENaC, sitio de entrada del litio a la célula.4
De manera fisiológica, las prostaglandinas antagonizan la acción de la ADH. Los antiinflamatorios no esteroideos (AINES), como indometacina, se han utilizado en el tratamiento de DI nefrogénica, debido a que inhiben la síntesis de prostaglandinas, de manera que se aumenta la acción de ADH. Su efecto neto puede tener una reducción de excreción urinaria de 25-50%, por lo que se puede adicionar a la terapia con tiazidas.14
DI CENTRAL
El manejo de la DI central se basa en cuatro principios 1) manejo del consumo de agua, 2) reemplazo hormonal de ADH con análogo sintético, 3) tratamiento de la causa de fondo, 4) seguimiento del tratamiento y vigilancia de la causa de fondo.15
En la mayoría de casos de DI central, el mecanismo osmorregulador de la sed está intacto y el consumo de líquido compensa adecuadamente las pérdidas renales. Sin embargo, cuando el paciente se ve sometido a diferentes situaciones como vómitos, puede llevar rápidamente a deshidratación hipernatrémica. Se debe hacer énfasis en los pacientes diagnosticados con DI de acudir a un centro médico en caso de vómitos o diarrea.15
La ADH es una hormona peptídica que tiene vida media en plasma de 5-10min. La desmopresina, un análogo de ADH se desarrolló para el tratamiento ambulatorio de DI actúa específicamente en los receptores V2 del riñón. Se diferencia de la hormona en dos maneras, se removió un grupo amino del amino ácido cisteína que prolongó la vida media a 6-8h. Además, se sustituye en la molécula la D-arginina por L-arginina, de manera que se pierde el efecto vasoconstrictor de la hormona, por lo que se puede usar libremente en pacientes con angina, sin riesgo del potencial vasoconstrictor. La presentación oral del medicamento es el más frecuente, en presentaciones de 100-400mcg.15
Se han utilizado otros medicamentos con menor frecuencia que aumentan la secreción de ADH o potencian su efecto a nivel renal, por ejemplo, clorpropamida, carbamazepina y AINES. Sin embargo, se han visto asociados con muchos efectos secundarios por lo que no se utilizan de manera generalizada.16
DI Transitoria y Enfermedad Grave por COVID19
La vasopresina se ha utilizado como medicamento vasopresor en pacientes gravemente enfermos, principalmente en unidades de cuidado intensivo (UCI). Se ha demostrado deficiencia de vasopresina en varios estados de shock, por lo que su uso está justificado en estados de shock séptico, posterior a cirugía cardiotorácica y para aumentar la presión arterial en casos de daño cerebral. Su acción en estos casos es a través del receptor V1 en el músculo liso de los vasos sanguíneos.17
Durante la actual pandemia del COVID19 se ha utilizado la vasopresina en UCI para pacientes críticamente enfermos con esta enfermedad. En el artículo “Transient diabetes insipidus in critically ill COVID19 patients” se describe una serie de 12 pacientes con internamiento en UCI que tuvieron diagnóstico de DI.17
El estudio observacional retrospectivo describe una serie de 152 pacientes internados en la UCI, de estos 37 pacientes fueron tratados con membrana de oxigenación extracorpórea veno-venosa (ECMO-VV). En 12 de estos pacientes se encontró 18 episodios de poliuria que fueron diagnosticados con DI. Ocho pacientes presentaron un único episodio, tres pacientes presentaron dos episodios y un paciente presentó cuatro episodios. Todos los episodios de DI transitoria menos uno, se presentaron posterior a descontinuar el tratamiento con vasopresina, con un tiempo promedio de instauración de DI de 5h posterior a descontinuarlo. El tratamiento con vasopresina tuvo una duración promedio de 2-5 días en los pacientes afectados.17
Previamente no se había descrito DI transitoria en pacientes sometidos a ECMO-VV y/o con diagnóstico de COVID19. Pese a que se necesita más investigación sobre el tema, es un posible efecto adverso que debe ser tomado en cuenta sobre el tratamiento con vasopresina.17
Conclusiones
La DI es una enfermedad que debido a su poca frecuencia y síntomas poco específicos resulta necesaria de estudiar y comprender. Su fisiopatología depende del tipo de DI que se presente, siendo en la central una secreción o producción insuficiente de ADH y en la nefrogénica una baja sensibilidad renal a la hormona. El principal reto diagnóstico es diferenciarla de polidipsia primaria. La medición de copeptina, un péptido co-secretado con la ADH ha aumentado la precisión diagnóstica del algoritmo clásico del test de deprivación de agua.
Para determinar la causa de DI se debe tomar en cuenta la presentación de los síntomas (aguda o gradual), así como síntomas y signos concomitantes que sugieran la causa de la enfermedad. Es necesario complementar con estudios adicionales dirigidos con base en la sospecha etiológica. El tratamiento debe ser dirigido dependiendo del tipo de DI identificada, y el tratamiento farmacológico se debe acompañar del no farmacológico para lograr un adecuado control de la enfermedad.
Conflicto de Interés
Los autores declaran no tener conflicto de interés.
Referencias bibliográficas
1. Christ-Crain M, Bichet DG, Fenske WK, Goldman MB, Rittig S, Verbalis JG, et al. Diabetes insipidus. Nature Reviews Disease Primers [Internet]. 2019;5(1):54. Available from: http://dx.doi.org/10.1038/s41572-019-0103-2
2. Christ-Crain M, Gaisl O. Diabetes insipidus. Presse Médicale [Internet]. 2021;50(4):1-11. Available from: http://dx.doi.org/10.1016/j.lpm.2021.104093
3. Refardt J, Winzeler B, Christ-Crain M. Diabetes insipidus: An update. Endocrinology Metabolism Clinics of North America [Internet]. 2020;49(3):517–31. Available from: http://dx.doi.org/10.1016/j.ecl.2020.05.012
4. Kavanagh C, Uy NS. Nephrogenic diabetes insipidus. Pediatric Clinics of North America [Internet]. 2019;66(1):227–34. Available from: http://dx.doi.org/10.1016/j.pcl.2018.09.006
5. Priya G, Kalra S, Dasgupta A, et al. Diabetes insipidus: A pragmatic approach to management. Cureus [Internet]. 2021;13(1):e12498. Available from: http://dx.doi.org/10.7759/cureus.12498
6. Bichet D. Clinical manifestations and causes of nephrogenic diabetes insipidus. Up to Date. 2021; 1-22
7. Hureaux M, Vargas-Poussou R. Genetic basis of nephrogenic diabetes insipidus. Molecular and Cellular Endocrinology [Internet]. 2023;560(111825):111825. Available from: http://dx.doi.org/10.1016/j.mce.2022.111825
8. Mutter CM, Smith T, Menze O, Zakharia M, Nguyen H. Diabetes insipidus: Pathogenesis, diagnosis, and clinical management. Cureus [Internet]. 2021;13(2):e13523. Available from: http://dx.doi.org/10.7759/cureus.13523
9. Bichet D. Clinical manifestations and causes of central diabetes insipidus. Up to Date. 2022; 1-21.
10. Christ-Crain M, Winzeler B, Refardt J. Diagnosis and management of diabetes insipidus for the internist: an update. Journal of Internal Medicine [Internet]. 2021;290(1):73–87. Available from: http://dx.doi.org/10.1111/joim.13261
11. Christ-Crain M. Diabetes insipidus: New concepts for diagnosis. Neuroendocrinology [Internet]. 2020;110(9– 10):859–67. Available from: http://dx.doi.org/10.1159/000505548
12. Garrahy A, Moran C, Thompson CJ. Diagnosis and management of central diabetes insipidus in adults. Clinical Endocrinology (Oxf) [Internet]. 2019;90(1):23–30. Available from: http://dx.doi.org/10.1111/cen.13866
13. Refardt J. Diagnosis and differential diagnosis of diabetes insipidus: Update. Best Practice & Research Clinical Endocrinology & Metabolism [Internet]. 2020;34(5):101398. Available from: http://dx.doi.org/10.1016/j.beem.2020.101398
14. Bichet D. Treatment of nephrogenic diabetes insipidus. Up to Date. 2021; 1-17.
15. Garrahy A, Thompson CJ. Management of central diabetes insipidus. Best Practice & Research Clinical Endocrinology & Metabolism [Internet]. 2020;34(5):101385. Available from: http://dx.doi.org/10.1016/j.beem.2020.101385
16. Bichet D. Treatment of central diabetes insipidus (vasopressin deficiency). Up to Date. 2021; 1-23.
17. Statlender L, Fishman G, Hellerman M, Kagan I, Bendavid I, Gorfil D, et al. Transient diabetes insipidus in critically ill COVID19 patients. Journal of Critical Care [Internet]. 2023;74(154211):154211. Available from: http://dx.doi.org/10.1016/j.jcrc.2022.154211
Versión Impresa
Esta obra está bajo una licencia internacional Creative Commons: Atribución-NoComercial-CompartirIgual 4.0 Internacional (CC BY-NC-SA 4.0)


Comments powered by CComment