Revisión bibliográfica
Enfermedad de Hirschsprung: una revisión de la literatura
Hirschsprung disease: a literature review
Dra. Valeria Caro Pizarro
Médico general
Licenciatura en Medicina y Cirugía, Universidad de Ciencias Médicas (UCIMED).
Médico trabajador independiente, San José, Costa Rica.
Miembro del Colegio de Médicos y Cirujanos de Costa Rica.
Costa Rica
Esta dirección de correo electrónico está siendo protegida contra los robots de spam. Necesita tener JavaScript habilitado para poder verlo.
Dra. Leyla Rockbrand Campos
Médico general, Licenciatura en Medicina y Cirugía, Universidad de Ciencias Médicas (UCIMED).
Médico interino de la Caja Costarricense del Seguro Social, San José, Costa Rica.
Miembro del Colegio de Médicos y Cirujanos de Costa Rica.
Costa Rica
Esta dirección de correo electrónico está siendo protegida contra los robots de spam. Necesita tener JavaScript habilitado para poder verlo.
Caro-Pizarro V, Rockbrand-Campos L. Enfermedad de Hirschsprung: una revisión de la literatura. Crónicas Científicas. Vol. 13. No. 13. Pág. 6-18. ISSN:2215-5171
Fecha de recepción: 10-04-2019
Fecha de aceptación: 18-06-2019
Resumen
La enfermedad de Hirschsprung es una patología congénita caracterizada por la disfunción del intestino y que se debe a la falla de migración de células embrionarias de la cresta neural o de la falla de la diferenciación de dichas células. Se clasifica en segmento corto, largo, ultracorto, colónica o intestinal, de acuerdo con la localización de la zona de transición entre el segmento aglangionico distal y el gangliónico proximal. Es la obstrucción intestinal más prevalente en pediatría, se presenta en 1:5000-7200 nacidos vivos, y predomina el sexo masculino. La mayoría de los casos ocurren de forma aislada y en minoría, aproximadamente 30 % se relacionan con malformaciones cromosómicas o alteraciones genéticas. Se manifiesta de forma característica por el estreñimiento inicial; otros síntomas son vómito, distensión abdominal, dolor a la palpación abdominal; la clínica puede variar de acuerdo con la edad de presentación. El diagnóstico se realiza mediante una biopsia rectal. El manejo terapéutico consiste en un abordaje médico previo a la realización de cirugía. Existen tres técnicas quirúrgicas principales, pero se adecuan de acuerdo con la extensión de la enfermedad o de las necesidades del paciente con el ideal de proporcionar mejor calidad de vida. Las complicaciones son variables, pero la enterocolitis es la más importante, pues implica la mayor mortalidad en dicha patología.
Palabras claves
Enfermedad de Hirschsprung, megacolon agangliónico congénito, manejo, diagnóstico.
Abstract
Hirschsprung disease is a congenital pathology characterized by functional bowel dysfunction, which is due to the failure of embryonic cell migration of the neural crest or the failure of the differentiation of these cells. It's classified in short, long, ultrashort, colonic or intestinal segment according to the location of the transition zone between the distal aganglionic segment and the proximal ganglionic segment. It's the most prevalent intestinal obstruction in pediatrics, occurs in 1:5000-7200 live births, and predominates male sex. Most cases are presented in isolation and in a minority, approximately 30 % is related to chromosomal malformations or genetic alterations. It's characteristically presented by initial constipation; other symptoms are vomiting, abdominal distention, abdominal palpation pain; the clinic may vary according to the age of presentation. The diagnosis is done by a rectal biopsy. Therapeutic management consists of a medical approach prior to the conduct of surgery. There are three main surgical techniques, but they are adapted according to the extension of the disease or patient needs with the ideal to provide a better life´s quality. The complications are variable, but the enterocolitis is the most important, since implies the highest mortality in this pathology.
Keywords
Hirschsprung's disease, congenital aganglionic megacolon, management, diagnosis.
Introducción
La enfermedad de Hirschsprung (EH), también conocida como megacolon agangliónico o congénito, fue descrita inicialmente por el pediatra Copenhague Harald Hirschsprung, en 1888, durante la observación clínica en dos lactantes que presentaron estreñimiento, hipertrofia y dilatación intestinal, pero no obstrucción mecánica (Carro, Ormaechea, Da Silva y Juambeltz, 2018; de Manueles, s. f.; Santos-Jasso, 2017). Al ser la constipación una de las causas de consultas frecuentes en la práctica pediátrica, se deben indagar sus posibles causas, entre ellas, la EH. Por lo tanto, desde hace dos siglos de su primera descripción, se ha desarrollado el tema hasta lograr reseñar manifestaciones clínicas, establecer pruebas diagnósticas y manejos terapéuticos de tipo médico como intervenciones quirúrgicas. Además, conociendo la evolución de dicha enfermedad, es posible entender mejor las complicaciones y el pronóstico que evidencian los pacientes. El objetivo de este artículo es realizar una revisión bibliográfica sobre la presentación clínica, el diagnóstico y el manejo terapéutico de dicha patología.
Metodología y materiales
Para el presente estudio, se efectuó una revisión bibliográfica de algunas fuentes electrónicas por medio de la búsqueda en bases de datos, específicamente: Sibdi, Binasss, SciELO, JAMA, Elsevier, Hospital Pediatrics, UpToDate Pediatrics in Review y PUBMED. En ellas, se indagaron diferentes publicaciones que trabajaban la enfermedad de Hirschsprung con un enfoque diagnóstico y de manejo. Para recolectar los artículos pertinentes para el desarrollo de este trabajo, se utilizaron ciertas palabras claves, tales como: enfermedad Hirschsprung, megacolon agangliónico congénito, estreñimiento. Se incluyeron artículos publicados entre los años 2010-2018, en inglés y español. Posterior a la recopilación, se integraron los datos provenientes de las fuentes escogidas, se revisaron y se organizó la información para exponerla en este documento.
Desarrollo
Definición
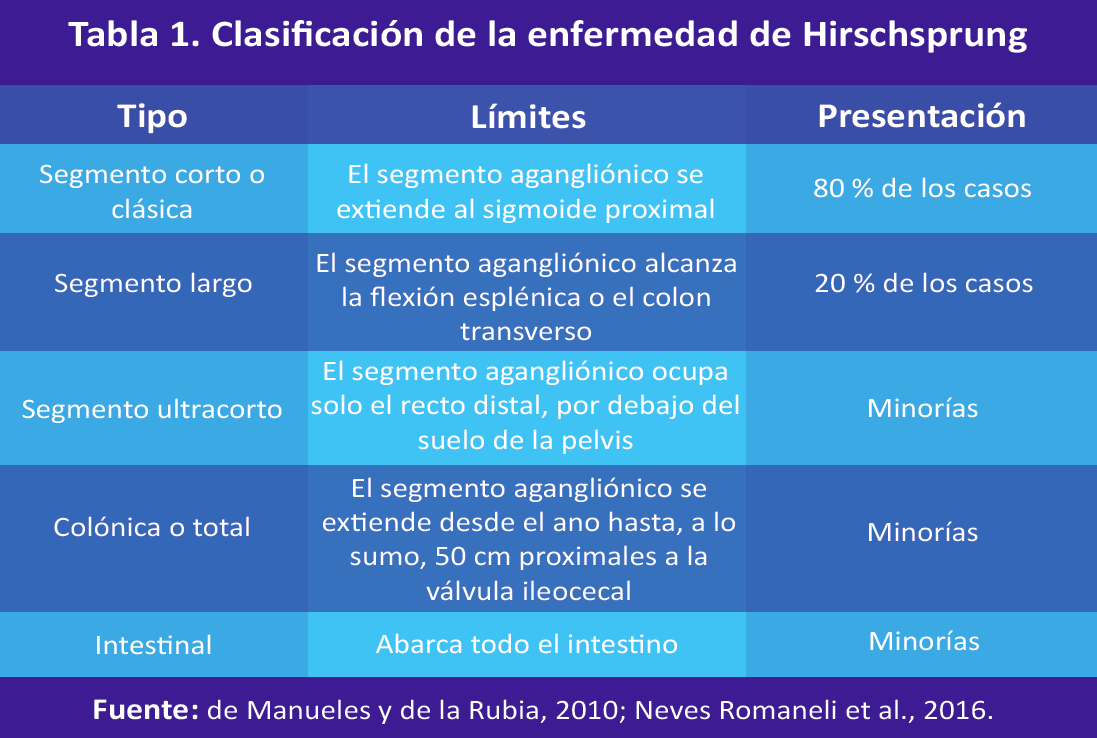
La EH es una patología congénita caracterizada por la disfunción funcional del intestino, debido a la falla de migración cefalocaudal en el tracto gastrointestinal de células embrionarias de la cresta neural o de la falla de la diferenciación de dichas células (Carro et al., 2018; de Manueles y de la Rubia, 2010; Neves Romaneli, Ribeiro, BustorffSilva, Carvalho y Lomazi, 2016). Se identifica por la presencia de un colon agangliónico de los plexos Meissner y Auerbach (García Arias y Ceciliano Romero, 2013).
Para clasificar la EH, se debe localizar anatómicamente el sitio de la transición entre el segmento aglangiónico distal y el segmento gangliónico proximal (Neves Romaneli et al., 2016) (ver Tabla 1). Además, el segmento agangliónico distal tiene como límite inferior el esfínter anal interno y el límite superior es variable (de Manueles y de la Rubia, 2010).
Incidencia
La incidencia de EH es 1:5000-7200 nacidos vivos, cuya mayor frecuencia se encuentra en el sexo masculino, con una relación hombre:mujer de 3:1 a 4:1 de ocurrencia; no tiene diferencia racial y es la causa más prevalente de obstrucción intestinal en pediatría (Msomi, Mangray y Du Plessis, 2017; Peyvasteh, Askarpour, Ostadian, Moghimi y Javaherizadeh,2016). Entre hermanos, la incidencia es de 3 % para el tipo segmento corto y hasta un 17 % para la clasificación segmento largo (Weesson y López, 2018).
La mayoría de los casos se da en los recién nacidos a término (RNT); una pequeña parte se presenta en niños mayores; en prematuros, su aparición es poco común; y en adultos, se han documentado alrededor de trescientos casos (Acurio, 2018; de Manueles, s. f.; López Ruiz et al., 2016; Weesson y López, 2018).
Genética
La EH aparece de forma aislada hasta en un 70 % de los casos, un 30 % a un 35 % aproximado de estos se asocian a malformaciones, de las cuales se puede observar hasta un 12 % en anomalías cromosómicas, y el restante, en otras anomalías (Carro et al., 2018; Chhabra y Kenny, 2016).
Según se ha estudiado, la malformación cromosómica que más se relaciona con la EH es la trisomía 21, síndrome de Down, cuyos casos tendrán cien veces más riesgo que la población general (Carro et al., 2018; Chhabra y Kenny, 2016). Cabe agregar que se estudia diariamente la conexión genética con esta patología, por lo que se han descrito numerosos genes en relación, por mencionar algunos: RET, GDNF, NTN, EDNRB, EDN3, ECE1, S0X10, ZFHX1B, PHOX2B, TCF-4, NTRK-3 (de Manueles y de la Rubia, 2010; Chhabra y Kenny, 2016). El gen que se vincula mayormente con EH es el gen Receptor tirosina quinasa (Ret), un protooncogén en el cromosoma 10q11, el cual se detecta en un 50 % en los casos familiares, y un 20 %, en los esporádicos (Chhabra y Kenny, 2016; de Manueles y de la Rubia, 2010).
Al ser una enfermedad causada por un fallo en el desarrollo embrionario, se han descrito casos con anomalías asociadas a fallos del sistema nervioso central. En ese mismo sentido, se deben considerar los factores externos contribuyentes, tales como: ambiental fetal, pirexia materna en el primer trimestre, que podrían llegar a contribuir al desarrollo de la enfermedad o anomalías asociadas (Chhabra y Kenny, 2016; de Manueles y de la Rubia, 2010).
Manifestaciones clínicas
La EH evidencia un síntoma característico inicial: el estreñimiento. Dependiendo del periodo de la vida, se definirá este síntoma de distintas formas; en el caso de los neonatos, es de aparición temprana y se determinará como la eliminación de meconio tardía; posterior a las 48 horas, en el caso de infantes, se caracteriza por deposiciones fecales esporádicas de una consistencia aumentada (de Manueles, s. f.; de Manueles y de la Rubia, 2010; Weber Estrada, 2012). Se debe considerar que la mayor parte de los RNT (99 %) tienen una eliminación de meconio previa a sus 48 horas de vida, pero en el caso de los prematuros tienden a postergar esta acción (Acurio, 2018; de Manueles, s. f.; de Manueles y de la Rubia, 2010).
A pesar de que el estreñimiento es un síntoma que debe levantar un alto grado de sospecha, no es un signo patognomónico. Se ha evidenciado que hasta un 40 % de los pacientes que se presentan con EH realizaron una eliminación de meconio en la primera hora de vida (Neves Romaneli et al., 2016).
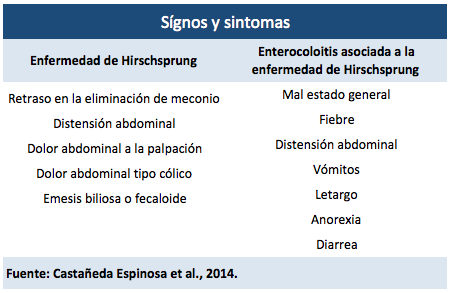
Otras manifestaciones clínicas que se pueden esperar en el paciente son síntomas de obstrucción intestinal distal, tales como retraso en la eliminación de meconio (el 95 % de los casos), distensión abdominal, dolor abdominal a la palpación y vómitos de contenido bilioso (de un 19 % a un 37 % de los casos) o fecaloide (Neves Romaneli et al., 2016; Weesson y López, 2018, 2019).
En algunos casos, cuando el diagnóstico no se realiza de forma temprana, el neonato manifestará enterocolitis asociada a la enfermedad de Hirschsprung (EAEH); el cuadro clínico se caracteriza por un mal estado general, fiebre, distensión abdominal, vómitos, letargo, anorexia y diarrea; inclusive, posterior a la realización del tacto rectal, puede presentar una explosión de heces gaseosas y líquidas (ver Tabla 2) (Weesson y López, 2018, 2019).
Tabla 2. Manifestaciones clínicas de la enfermedad de Hirschsprung y enterocolitos asociada a la enfermeddad de Hirschsprung.
En el niño entre los 3 años y los 14 años de edad, el desarrollo de EH se caracteriza por estreñimiento crónico severo, asociado a un abdomen con fecalomas palpables, un esfínter hipertónico, ampolla rectal con heces duras y, usualmente, presenta un déficit nutricional (Acurio, 2018; de Manueles, s. f.).
Las manifestaciones clínicas en el adulto son menos complejas, ya que durante su vida ha desarrollado una compensación: el segmento previo a la zona aganglionar sufre hipertrofia, lo que ayuda a contrarrestar la obstrucción mecánica (López Ruiz et al., 2016).
Diagnóstico
La sospecha de la presencia de la EH se debe basar en las características clínicas expuestas anteriormente, además de complementarlas con estudios de imagen contrastados o manometrías. Para establecer el diagnóstico, es necesario realizar una biopsia rectal (Msomi et al., 2017; Santos-Jasso, 2017; Weesson y López, 2018).
El estudio de imagen de preferencia para ayudar al diagnóstico es el colon por enema, en el cual se busca una zona estrecha, sin peristaltismo, y proximal a esta zona se localiza la dilatación intestinal, conocida como zona de transición; por lo general, se encuentra en el rectosigmoides (Carro et al., 2018; Santos-Jasso, 2017). Aunque, se ha observado que la zona de transición definida macroscópicamente de manera radiológica no concuerda con la que se determina histopatológicamente; un 12 % de los casos aproximadamente y de predominio sí está localizada en el rectosigmoides (Santos-Jasso, 2017). Además, se ha notado que en el 75 % de los niños con EH total no se encuentra la zona de transición (Santos-Jasso, 2017). La sensibilidad de este estudio en excluir la enfermedad es de un 80 % al 88 %, aproximadamente (Msomi et al., 2017).
Por su parte, la manometría anorrectal traduce la ausencia de la relajación involuntaria del esfínter anal interno, con una sensibilidad del 91 % y una especificidad del 94 %. Cuando la materia fecal se localiza en la ampolla rectal, se activa el reflejo de relajación de esfínter interno, el cual se encuentra ausente cuando no hay células ganglionares; este procedimiento se utiliza más como una prueba de tamizaje, por lo que no confirma el diagnóstico (Santos-Jasso, 2017).
La biopsia rectal es el gold standard para el diagnóstico de EH, pues significa un 93 % de sensibilidad y un 98 % de especificidad (Santos-Jasso, 2017). El estudio se puede efectuar de dos maneras; se recomienda que, de forma inicial, se realice una biopsia por aspiración a 3 cm de la línea dentada, para evitar la zona que fisiológicamente es agangliónica, adquiriendo tejido mucoso y submucosa; también se aconseja tomar una segunda muestra proximal a la primera (Santos-Jasso, 2017; Weesson y López, 2018). La biopsia por aspiración se puede realizar al pie de la cama del paciente o de forma ambulatoria y sin anestesia general (Carro et al., 2018; Santos-Jasso, 2017; Weesson y López, 2018). Cuando se obtiene un resultado patológico no concluyente o de tejido insuficiente, se sugiere la realización de una segunda biopsia por succión o de espesor total, esta última requiere anestesia general en sala de operaciones(9).
La biopsia determina si las células ganglionares están ausentes en el plexo submucoso, resultado determinante para concluir en el diagnóstico (Carro et al., 2018; Santos-Jasso, 2017; Weesson y López, 2018). La muestra se somete a una tinción hematoxilina y eosina (Carro et al., 2018; Santos-Jasso, 2017). No obstante, algunos casos requieren confirmación del diagnóstico, por lo que en la muestra se buscan otros hallazgos y se le efectúan pruebas adicionales; por ejemplo, inmunohistoquímica para células ganglionares con proteína S-100, presencia de fibras nerviosas hipertróficas, aumento de la actividad de la acetilcolinesterasa o tinción en la mucosa muscular, y disminución o ausencia de calretinina (ver Figura 1) (Carro et al., 2018; Gunadi, Karina y Dwihantoro, 2018; Santos-Jasso, 2017; Weesson y López, 2018).
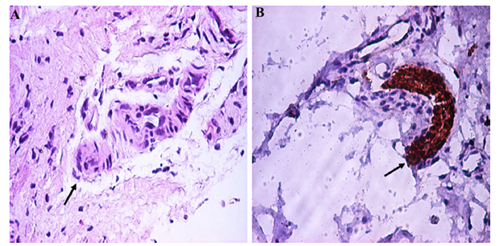
Figura 1. Hallazgos histopatológicos de la biopsia rectal de espesor total en un paciente con enfermedad de Hirschsprung.
Nota: La flecha muestra un tronco nervioso hipertrófico y la ausencia de células ganglionares. La imagen A indica una tinción de hematoxilina y eosina (x200). Lla imagen B, una inmunohistoquímica S100 (x200).
Fuente: Copyright 2018 BMC Research Notes.
Diagnóstico diferencial
La EH es una patología que, por sus complicaciones, puede provocar la muerte del paciente. Por esa razón, un diagnóstico oportuno es esencial. Es importante considerar que sus síntomas se asemejan a los de otras enfermedades (ver Tabla 3) (de Manueles y de la Rubia, 2010).
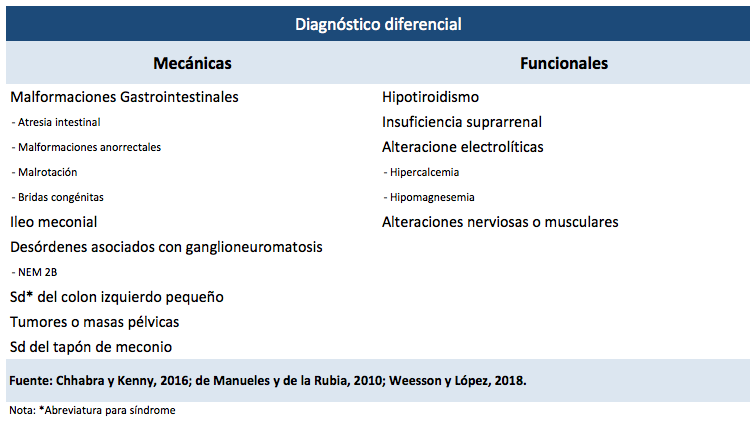
Una de las más importantes es la neurodisplasia intestinal (NID), la cual se caracteriza por anomalías en los plexos mientéricos y submucosos, que presentan una formación errónea de los ganglios, un aumento cuantitativo de las células ganglionares, una hiperplasia de plexos mesentéricos y una actividad acetilcolinesterasa (AchE) positiva (Acurio, 2018; Arnez Durán y Villarroel Gutiérrez, 2014).
También es posible dividir los diagnósticos diferenciales entre los que muestran un cuadro obstructivo funcional y los que ofrecen uno obstructivo mecánico (Weesson y López, 2018).
Dentro de los ocasionantes del estreñimiento mecánico, se encuentran: a) las malformaciones gastrointestinales, por ejemplo, atresia intestinal, malformaciones anorrectales, malrotación y bridas congénitas; b) el íleo meconial por fibrosis quística; c) desórdenes asociados con ganglioneuromatosis (incluido el NEM 2B); d) síndrome del colon izquierdo pequeño; e) tumores o masas pélvicas; y f) síndrome del tapón de meconio (Chhabra y Kenny, 2016; de Manueles y de la Rubia, 2010; Weesson y López, 2018).
En el caso de las enfermedades que provocan un estreñimiento funcional, existen: a) hipotiroidismo; b) insuficiencia suprarrenal; c) alteraciones electrolíticas del potasio, por ejemplo, hipercalcemia o hipermagnesemia; y en raras alteraciones nerviosas o musculares (Chhabra y Kenny, 2016; de Manueles y de la Rubia, 2010).
Cabe mencionar que la EH clásica debe ser diferenciada de EH de segmento ultracorto, pero en este caso se presentará de una forma más leve, tiene menos tendencia a desarrollar complicaciones, como la enterocolitis y el retraso en el crecimiento (Weesson y López, 2018).
Manejo terapéutico
El tratamiento de la EH busca mejorar la calidad de vida del paciente, restablecer su continuidad digestiva y evitar o disminuir las complicaciones.
Para cumplir con este objetivo, las técnicas quirúrgicas han evolucionado hasta lograr disminuir la morbilidad y la mortalidad de los pacientes (Acurio, 2018; de Manueles, s. f.; Santos-Jasso, 2017); este avance es relevante, pues después del establecimiento del diagnóstico, se busca una resolución principalmente quirúrgica (de Manueles, s. f.).
Manejo médico
El manejo médico se basa en medidas de reanimación iniciales, comenzando por la hidratación del paciente para terminar con la preparación previa a la cirugía (Chhabra y Kenny, 2016; Weesson y Lopez, 2019). El primer paso es valorar la hidratación del paciente y, de acuerdo con esto, administrar fluidos intravenosos (IV); el segundo paso consiste en la colocación de una sonda nasogástrica (SNG), que permitirá la descompresión gástrica; esta debe ser aspirada con regularidad (Chhabra y Kenny, 2016; Weesson y Lopez, 2019).
También es necesario administrar antibióticos de amplio espectro, como metronidazol y vancomicina, para evitar la translocación bacteriana y la enterocolitis. Además, se debe realizar un estimulación rectal con un tubo rectal de 10 o 12 French; esto, al igual que el tacto rectal, puede provocar la salida explosiva de heces (Chhabra y Kenny, 2016; Weesson y Lopez, 2019).
Se necesita una preparación del tracto gastrointestinal previa a la intervención de la cirugía. Para ello, se hará una evacuación de todas las heces por medio de enemas evacuantes y la estimulación rectal, con el fin de prevenir complicaciones conocidas como la enterocolitis (de Manueles y de la Rubia, 2010; Weber Estrada, 2012).
Manejo quirúrgico
El abordaje quirúrgico ha cambiado a lo largo de los años. Inicialmente, se empleaban procedimientos invasivos en varios tiempos; ahora, se ha cambiado a los mínimamente invasivos en un solo tiempo, reduciendo así las complicaciones asociadas. No obstante, la decisión de un abordaje de este tipo dependerá del cirujano, de la edad del paciente, del estado nutricional de este, de la longitud de la zona aganglionar, de la longitud y la reversibilidad de la dilatación cólica (Chhabra y Kenny, 2016; García Arias y Ceciliano Romero, 2013; López Ruiz et al., 2016; Weesson y López, 2018). Aun así, todas las técnicas quirúrgicas tienen como meta resecar la zona aganglionar.

Figura 2. (A) Anatomía preoperatoria. (B) Técnica de Swenson. (C) Procedimiento de Duhamel. (D) Procedimiento de Soave.
Fuente: Copyright 2016 Surgery (Oxford).
Existen variaciones en el abordaje quirúrgico dependiendo de la presentación de la enfermedad. Por ejemplo, al enfrentarse a un caso de EH total, la técnica generalmente utilizada es la de Martín. Este procedimiento consiste en una anastomosis latero-lateral al íleo y se conserva una parte del colon agangliónico, por lo cual esta técnica permite explotar la capacidad absortiva del colon aganglionar y la potencia motriz del intestino delgado (Acurio, 2018). En el caso de un paciente con EH de segmento ultracorto, se puede realizar la miomectomía, cuyo proceso se ejecuta vía transanal, resecando la región superior del esfínter anal interno (López Ruiz et al., 2016).
Cabe agregar que la necesidad de disminuir la morbilidad, las complicaciones y los costos hospitalarios han servido como motor para desarrollar técnicas mínimamente invasivas; por ejemplo, la cirugía laparoscópica (Chhabra y Kenny, 2016; de Manueles, s. f.). Este método quirúrgico ayuda a la movilización del colon proximal hacia el rectosigmoides y se preforma en conjunto con la resección transanal; al llevar a cabo este procedimiento, es importante reconocer y conservar la línea dentada para lograr una preservación de la sensibilidad y de la lubricación a largo plazo (Chhabra y Kenny, 2016; de Manueles, s. f.).
No obstante, los resultados a largo plazo entre las técnicas nuevas y las clásicas son semejantes (de Manueles y de la Rubia, 2010; Weesson y López, 2018). Además, todos los procedimientos quirúrgicos pueden asociarse a colostomías e ileostomías previas (Santos-Jasso, 2017). De la Torre, en 1998, desarrolló una técnica que se asemeja a la desarrollada por Soave, pero en este caso se realiza por vía transanal; esta ha ido ganando una gran aceptación en los últimos años (de Manueles y de la Rubia, 2010).
Complicaciones
La enterocolitis es la complicación más importante y es la causante de la mortalidad en el desarrollo de la EH. Según la literatura, su incidencia se encuentra alrededor del 5 % al 45 % y, usualmente, se presenta en el primer año posquirúrgico (Chhabra y Kenny, 2016; de Manueles y de la Rubia, 2010). Los pacientes que tienen más riesgo de manifestar esta complicación son los diagnosticados con EH de segmento largo, también se encuentra asociada a pacientes con trisomía 21, déficit nutricional, estenosis anastomóticas (Chhabra y Kenny, 2016; de Manueles y de la Rubia, 2010; Weesson y López, 2018). Su tratamiento consiste, principalmente, en la descompresión intestinal, la administración de fluidos de manera intravenosa y el suministro de antibióticos de amplio espectro para asegurar la cobertura de gramnegativos (Chhabra y Kenny, 2016).
Otra de las complicaciones comúnmente exhibida es la incontinencia fecal, que se define como la imposibilidad total o parcial de controlar voluntariamente la expulsión de gases y material fecal (Carro et al., 2018). Sucede de manera habitual durante el posoperatorio temprano, con hasta diez deposiciones diarias en los días consecutivos a la cirugía (de Manueles y de la Rubia, 2010; Weesson y López, 2018). Se puede deber a la disfunción del esfínter y a la disminución de la superficie absortiva del colon (Acurio, 2018; de Manueles y de la Rubia, 2010; Weesson y López, 2018).
En un estudio desarrollado por Carro (2018), se documentó que dependía de la técnica utilizada para saber el porcentaje de los pacientes que presentan incontinencia; por ejemplo, al emplear la técnica de De la Torre, un 75 % presentó esta complicación de forma leve, en cambio, con la técnica Soave ocurrió solamente en un 20 %. No obstante, seis meses después de la cirugía, esta complicación muestra una mejoría significativa en la mayoría de los pacientes (75 % al 95 %) (de Manueles y de la Rubia, 2010; Weesson y López, 2018).
El estreñimiento forma parte del espectro de patologías donde la EH representa un importante factor de riesgo, puede llegar a afectar del 10 % al 30 % de los pacientes, siendo más común en los primeros meses posquirúrgicos. También se asocia más a las técnicas de Duhamel, debido a que estas conservan más zonas de tejido aganglionar (Chhabra y Kenny, 2016; de Manueles y de la Rubia, 2010; Weesson y López, 2018). Respecto del tratamiento, un manejo conservador con enermas y laxantes orales representa la primera opción terapéutica; en aquellos casos donde existe persistencia de los síntomas, se inician otros estudios complementarios, tales como biopsia, enema opaco y manometría; posteriormente, se recurre a manejos más invasivos, como dilataciones forzadas, inyecciones de toxina botulínica o revisión quirúrgica (de Manueles y de la Rubia, 2010).
Por último, se encuentra la enuresis, una de la complicaciones menos frecuentes, secundaria a daño a la inervación pélvica o a neuropatías. La forma idónea para disminuir su incidencia es por medio de la técnica laparoscópica (de Manueles y de la Rubia, 2010).
Conclusiones
La EH es una patología congénita que presenta un colon agangliónico de los plexos Meissner y Auerbach, que se produce por la falla de migración de células de la cresta neural o de la falla de la diferenciación de dichas células. Para clasificar la enfermedad según su extension se debe localizar la zona de transición; el caso más común es el segmento corto (80 %). La mayoría de los casos ocurren en RNT y es más frecuente en hombres. De un 30 % a un 35 % de estos se asocia a malformaciones cromosómicas y anomalías genéticas. La malformación cromosómica más frecuente es la trisomía 21. El gen con mayor vinculación es RET y se detecta en un 50 % en casos familiares y en un 20 % en los esporádicos. El síntoma característico es el estreñimiento. Si el diagnóstico no se realiza de forma temprana, el neonato puede presentar EAEH. El manejo médico de la enfermedad se debe enfocar en la hidratación y en la desimpactación del paciente. El abordaje quirúrgico depende de la extensión de la enfermedad; las principales técnicas son Swenson, Duhamel y Soave. La cirugía laparoscópica ha demostrado disminuir la morbilidad, las complicaciones y los costos hospitalarios. Los resultados a largo plazo entre las técnicas nuevas y las clásicas son semejantes. La EAEH es la complicación más severa, pues presenta alta mortalidad si no es abordada a tiempo.
Referencias bibliográficas
Acurio, D. (2018). Análisis de un caso clínico de Enfermedad de Hirschsprung un paciente pediátrico del Hospital General Latacunga, 2018. (1.ª ed.). Ambato, Ecuador: Universidad Regional Autónoma de los Andes - UNIANDES.
Arnez Durán R. y Villarroel Gutiérrez, M. (2014). Neurodisplasia Intestinal: un caso inusual de distensión abdominal. Gaceta Médica Boliviana, 37(2), 94-96.
Carro, G., Ormaechea, M., Da Silva, E. y Juambeltz, C. (2018). Enfermedad de Hirschsprung: resultados del tratamiento quirúrgico en el Hospital Pediátrico Pereira Rossell. Archivos De Pediatría Del Uruguay, 89(3), 158-164. doi: 10.31134/ap.89.3.2
S. Castañeda Espinosa, A. García Giraldo, P. Jaimes de la Hoz, L. Jaramillo Barberi*, M.A. Perilla López, M. Méndez Manchola, J.A. Niño Salcedo, F. Fierro Ávila (2014). Enterocolitis asociada a enfermedad de Hirschsprung. Experiencia en un Hospital Universitario Pediátrico. Revista Cirugía Pediátrica, 27(2), 78-83.
Chhabra, S. y Kenny, S. (2016). Hirschsprung's disease. Surgery (Oxford), 34(12), 628-632. doi: 10.1016/j.mpsur.2016.10.002
de Manueles, J. (s. f.). Enfermedad de Hirschprung. Protocolos diagnósticos y terapéuticos en pediatría. (1.ª ed.). Madrid: Sociedad Española de Pediatría.
de Manueles, J. y de la Rubia Fernández, L. (2010). Enfermedad de Hirschprung. Protocolos diagnóstico y terapéuticos de gastroenterología, hepatología y nutrición pediátrica. (2.ª ed.). Madrid: Ergón S. A.
García Arias, F. y Ceciliano Romero, N. (2013). Análisis del manejo quirúrgico de la enfermedad de Hirschsprung en el Hospital Nacional de Niños Dr. Carlos Sáenz Herrera, durante el periodo 2000-2010. Acta Médica Costarricense, 55(2), 87-91.
Gunadi, Karina, S. y Dwihantoro, A. (2018). Outcomes in patients with Hirschsprung disease following definitive surgery. BMC Research Notes, 11(1), 1-5. doi: 10.1186/s13104-018-3751-5
López Ruiz, J., Tallón Aguilar, L., Sánchez Moreno, L., López Pérez, J., Pareja Ciuró, F., Oliva Mompeán, F. y Padillo Ruiz, F. (2016). Hirschsprung disease with debut in adult age as acute intestinal obstruction: case report. Revista Española De Enfermedades Digestivas, 108(11), 742-746. doi: 10.17235/reed.2016.3841/2015
Msomi, M., Mangray, H. y Du Plessis, V. (2017). An assessment of the accuracy of contrast enema for the diagnosis of Hirschsprung disease at a South African tertiary hospital. South African Journal of Radiology, 21(1), 1-5. doi: 10.4102/sajr.v21i1.1093
Neves Romaneli, M., Ribeiro, A., Bustorff‐Silva, J., Carvalho, R. y Lomazi, E. (2016). Doença de Hirschsprung – Dismotilidade intestinal pós‐ cirúrgica. Revista Paulista De Pediatria, 34(3), 388-392. doi: 10.1016/j.rpped.2015.12.008
Peyvasteh, M., Askarpour, S., Ostadian, N., Moghimi, M. y Javaherizadeh, H. (2016). Diagnostic Accuracy Of Barium Enema Findings In Hirschsprung's Disease. ABCD. Arquivos Brasileiros De Cirurgia Digestiva (São Paulo), 29(3), 155-158. doi: 10.1590/0102-6720201600030007
Santos-Jasso, K. (2017). Enfermedad de Hirschsprung. Acta Pediátrica De México, 1(1), 72-78. doi: 10.18233/apm1no1pp72-781325
Weber Estrada, N. (2012). Enfermedad de Hirschsprung. Revista Médica de Costa Rica y Centroamérica, LXIX(602), 251-256.
Weesson, D. y López, M. (2018). Congenital aganglionic megacolon (Hirschsprung disease). UpToDate. Recuperado de https://www.uptodate.com/contents/congenital-aganglionic-megacolon-hirschsprung-disease
Weesson, D. y Lopez, M. (2019). Emergency complications of Hirschsprung disease. UpToDate. Recuperado de https://www.uptodate.com/contents/emergency-complications-of-hirschsprung-disease
Versión Impresa
Esta obra está bajo una licencia internacional Creative Commons: Atribución-NoComercial-CompartirIgual 4.0 Internacional (CC BY-NC-SA 4.0)


Comments powered by CComment